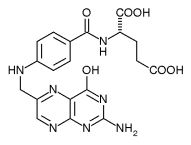
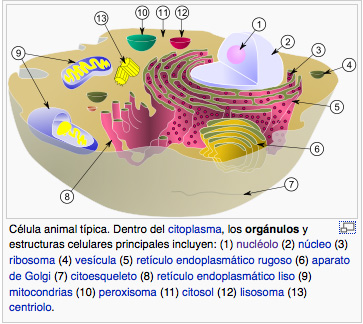
Se publica el primer estudio sobre la mutación de genes asociados al Parkinson en pacientes extremeños
El primer estudio que se realiza en Extremadura relativo a la prevalencia de mutaciones en ciertos genes asociados a la enfermedad de Parkinson en una muestra de pacientes de la comunidad autónoma acaba de ser publicado, según informa la Universidad de Extremadura (UEx) y recoge Europa Press. Se trata de un proyecto del Grupo de […]
Read More »